

Introduction: The 20q deletion [del(20q)] is a recurrent chromosomal aberration in myelodysplastic syndromes (MDS) and, as a single abnormality, is associated according to the Revised International Prognostic Scoring System (IPSS-R) with a favorable outcome. However, the breakpoint of del(20q) is very heterogeneous and may cause deletion of the ASXL1 gene (20q11.21). This gene is an important epigenetic regulator of hematopoiesis and its mutations have been associated in MDS with a shorter overall survival (OS) and a lower response to azacitidine (AZA).
Aim: To assess the incidence, prognostic value and impact on response to AZA of ASXL1 chromosomal alterations and genetic mutations in MDS patients with del(20q).
Methods: We studied 153 patients diagnosed with MDS and del(20q) as a sole abnormality (n=93), with an additional chromosomal abnormality (n=27) or in a complex karyotype (n=33). Response to AZA therapy was assessed using the 2006 International Working Group (IWG) criteria. Analysis of ASXL1 chromosome alterations was performed by FISH (Empire Genomics probe). Samples with ASXL1 alterations by FISH were analyzed using the Agilent SurePrint G3 Human CGH 8x60K Microarray. Mutations of ASXL1 were detected by Sanger sequencing. SF3B1, SRSF2, U2AF1, DNMT3A, IDH1, IDH2, TP53, RUNX1, and SETBP1 mutations were screened by high-resolution melting and positives were confirmed by Sanger sequencing. An in vitro assay of the response to AZA in HAP1 and HAP1 ASXL1 knockout cell lines (Horizon) was also performed. The association of the clinical characteristics with the molecular findings was analyzed with the SPSS statistical program (v.20.0) and P values <0.05 were considered as statistically significant.
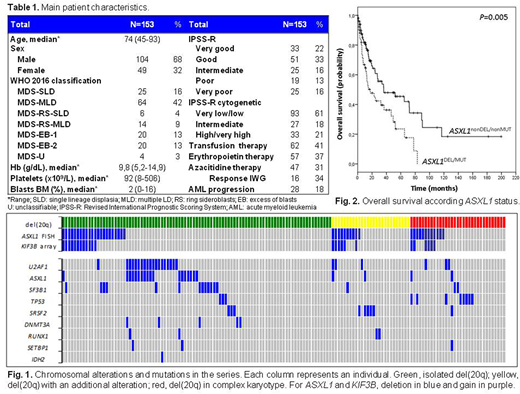
Results: Main patient characteristics are shown in Table 1.ASXL1 chromosomal alterations were detected by FISH in 44 patients (29%; Fig. 1): 34 patients (22%) with gene deletion (ASXL1DEL) and 10 cases (6.5%) with additional gene copies (ASXL1GAIN). All 44 cases were analyzed by microarray, and alterations in centromeric KIF3B gene (20q11.21) were also identified in 24 patients (16%). Patients with ASXL1DEL showed a lower platelet count (median, 68x109/L vs. 103x109/L, P=0.046), a poorer response to AZA (response, 9% vs. 43%, P= 0.040) and a trend towards a lower OS (34 vs. 65 months, P=0.057). ASXL1 and KIF3B chromosomal gains were associated with complex karyotypes (ASXL1GAIN, 80% vs. 23%, P<0.001; KIF3BGAIN, 80% vs. 25%, P=0.011). Patients with ASXL1GAIN had a similar OS and response to AZA than ASXL1nonDEL/ASXL1nonMUT patients (OS, 51 vs. 73 months, P=0.86; response, 60% vs. 44%, P=0.43). Seventy patients (46%) in the series had ≥1 genetic mutation (Fig. 1). ASXL1 mutations (ASXL1MUT; 22/153, 14%) were mostly frameshift (n=17, 77%) or nonsense (n=4, 18%) and were associated with a lower hemoglobin level (median, 8.3 vs. 10.3 g/dL; P=0.007), a higher ferritin level (median, 691 vs. 286 ng/mL, P=0.014), a higher number of mutations (23% vs. 0% ≥3 mutations, P<0.001) and a lower OS (22 vs. 62 months, P=0.016). U2AF1 mutations were associated with a deeper neutropenia (median, 1.17x109/L vs. 1.83x109/L, P=0.026) and TP53 mutations were associated with acute myeloid leukemia (AML) transformation (55% vs. 16%, P=0.005) and a lower OS (24 vs. 57 months, P=0.037). In the global series, patients with ASXL1 altered either by chromosomal deletion or mutation (ASXL1DEL/ASXL1MUT) had a poorer response to AZA (13% vs. 47%, P=0.020) and a lower OS (32 vs. 70 months, P=0.005; Fig. 2) than the remaining patients. In the analysis of isolated del(20q) subgroup (n=93), ASXL1DEL/ASXL1MUT patients also showed a lower OS (37 vs. 80 months, P=0.026) and ASXL1DEL was associated with a higher risk of progression to AML (33% vs. 11%, P=0.029), despite 64% of patients were clustered in lower-risk categories of the IPSS-R. In the in vitro assay performed at 2 μM AZA, HAP1 ASXL1 knockout cells showed less growth inhibition (26% vs. 46%, P=0.003), lower cell apoptosis (17% vs. 34%, P<0.001) and less cell cycle arrest in G0/G1 phase (6% vs. 23%, P=0.009), compared to HAP1 cells.
Conclusion: In MDS patients with del(20q), the alteration of ASXL1 by chromosome deletion or somatic mutation was associated with adverse clinical features and a poorer response to AZA. Its detection at diagnosis would allow the rapid identification of a subgroup of MDS patients with a poor prognosis that could benefit of earlier and more effective therapies.
Sanz:Abbvie Pharmaceuticals: Membership on an entity's Board of Directors or advisory committees; LaHoffman Roche Ltd.: Membership on an entity's Board of Directors or advisory committees; Helsinn: Membership on an entity's Board of Directors or advisory committees; Takeda Pharmaceutical Ltd.: Membership on an entity's Board of Directors or advisory committees. Tormo:Novartis: Honoraria, Membership on an entity's Board of Directors or advisory committees; Janssen: Honoraria; Celgene: Honoraria, Membership on an entity's Board of Directors or advisory committees; Pfizer: Honoraria; MSD: Honoraria; Daiichi Sankyo: Honoraria; Servier: Honoraria; Roche: Membership on an entity's Board of Directors or advisory committees; Astellas: Membership on an entity's Board of Directors or advisory committees.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal